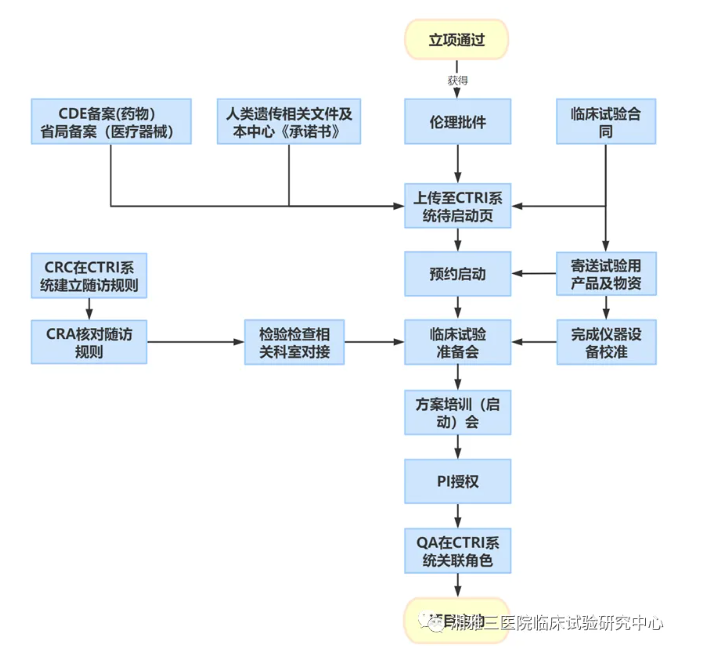
1.项目通过我院医学伦理委员会批准;临床试验合同签署完成;涉及人类遗传资源的根据《人类遗传资源管理条例》获得备案或审批,完成本中心人类遗传资源《承诺书》签字,并在科学技术部政务服务平台完成备案;药物临床试验在CDE备案登记;医疗器械临床试验在申办者所在地省、自治区、直辖市食品药品监督管理部门完成备案;并在CTRI系统待启动页面上传上述相关文件,预约方案培训时间。
2.启动前,CRC在CTRI系统录入随访规则,CRA进行核对。CRC协助研究者与临床试验检验检查涉及的相关科室完成对接。
3.启动前,CRA确认研究物资(试验用药品、文件资料、相关仪器设备)到达中心,确认试验相关仪器设备获得校准,取得校准证书。
4.启动前,机构办公室QA组织项目主要负责医生、CRA、CRC召开准备会,机构办公室QA再次与CRA或CRC确认临床试验用药品、资料、试验物资、检查检验相关科室对接情况、CTRI系统临床试验项目录入情况。共同讨论项目具体筛选、入选流程、实验室检查、过程中各种工具表格设计与应用、药品管理、生物样本管理等各项内容。机构办公室QA记录《临床试验准备会记录表》并存档。
5.启动前准备工作完成后,申办者/CRO与机构办公室、项目PI协商后确定临床试验启动时间、地点、内容及其他相关安排。
6.在机构办公室的统一组织和协调下,组织专业科室开展试验方案、标准操作规程(SOPs)、知情同意、AE和SAE、原始数据记录、CRF/eCRF、合并用药及相关法规的学习和培训。项目PI、研究医生、研究护士、药品保管人员、机构办公室人员必须到会并签到。必要时请负责试验相关检验、检查科室的人员参加。参与启动会人员针对培训内容和试验操作展开讨论。
7. 项目PI在试验开展前完成任务授权分工表,需要保证有足够的人力、物力来完成临床试验。授权完成后,机构办公室QA根据《授权分工表》在CTRI系统关联相关角色。
8. 启动会完成后,申办方/CRO对于启动会的培训内容,答疑及未解决问题进行记录,会后逐条回复至研究者。同时,机构办公室及监查员应拟定QA和监查计划,确保项目顺利开展。